Lab 4: Homology & Phylogeny
Phylogenetic Tree
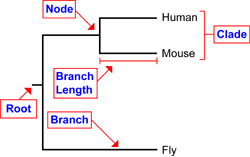
Node: represents a taxonomic unit. This can be either an existing species or an ancestor.
Branch: defines the relationship between the taxa in terms of descent and ancestry.
Topology: the branching patterns of the tree.
Branch length: represents the number of changes that have occurred in the branch.
Root: the common ancestor of all taxa.
Distance scale: scale that represents the number of differences between organisms or sequences.
Clade: a group of two or more taxa or DNA sequences that includes both their common ancestor and all of their descendents.
Branch: defines the relationship between the taxa in terms of descent and ancestry.
Topology: the branching patterns of the tree.
Branch length: represents the number of changes that have occurred in the branch.
Root: the common ancestor of all taxa.
Distance scale: scale that represents the number of differences between organisms or sequences.
Clade: a group of two or more taxa or DNA sequences that includes both their common ancestor and all of their descendents.
What you will do today
| mega_lab_2-13-24.docx |
Install MEGA on either your MAC or PC:
STEP 1: Multiple Sequence Alignment
SAMPLE MEGA files: https://www.megasoftware.net/examples
Next steps are for just FASTA files (Convert FASTA to MEGA files):
STEP 2: Build a tree in MEGA
10. Click on the Phylogeny button in MEGA
11. Select which type of method you want to choose (Neighbor joining or Maximum likelihood—try both)
11. Upload your Multiple sequence alignment file from your desktop (Gene NameAligment.meg)
12. Make sure you select BOOTSTRAP method in the window that pops up
13. Export tree as a PNG file: IMAGE> Save as PNG name it your gene+Method.png
*note maximum likelihood trees will take a long time to build, Neighbor joining is faster
*BOOTSTRAPPING: In terms of your phylogenetic tree, the bootstrapping values indicates how many times out of 100 (in your case) the same branch was observed when repeating the phylogenetic reconstruction on a re-sampled set of your data.
How to build trees using MEGA (by Max Haase '17): http://haasegen564s17.weebly.com/mega.html (note old version of MEGA used) *if you are looking for example files to use in MEGA you can find these files HERE. Try the CRAB rRNA file as an Example.
Alternative ways to build a tree:
Align using Clustal Omega: https://www.ebi.ac.uk/Tools/msa/clustalo/
Then paste the alignment into:
Simple Phylogeny: https://www.ebi.ac.uk/Tools/phylogeny/simple_phylogeny/
- MAC: Download MEGA X (https://www.megasoftware.net/) not MEGA 11
- Windows; Only option is MEGA 11
STEP 1: Multiple Sequence Alignment
- Go to Entrez Gene or Ensemble to retrieve PROTEIN homologs (Ensemble can download .MEG files so it’s faster). Homologene you can download a FASTA file and convert to a MEG file in MEGA.
SAMPLE MEGA files: https://www.megasoftware.net/examples
Next steps are for just FASTA files (Convert FASTA to MEGA files):
- Export FASTA files for each model organisms to your desktop (name Human.fasta, etc).
- Click on your Human FASTA file. It should open MEGA if you have MEGA installed.
- Click Align File
- EDIT> Insert Sequences from File >
- They should all be in MEGA now
- To do MULTIPLE sequence alignment you have 2 methods: Clustal W or Muscle (you can try both). Click on the W in the toolbar to align using Clustal W or Click on the Arm icon to use Muscle.
- Now your sequences are aligned in MEGA to be used for the next step. USE the scroll bar to see the alignments
- In toolbar on top: Select DATA>Export file as MEGA format (name Gene name Alignment.meg)
STEP 2: Build a tree in MEGA
10. Click on the Phylogeny button in MEGA
11. Select which type of method you want to choose (Neighbor joining or Maximum likelihood—try both)
11. Upload your Multiple sequence alignment file from your desktop (Gene NameAligment.meg)
12. Make sure you select BOOTSTRAP method in the window that pops up
13. Export tree as a PNG file: IMAGE> Save as PNG name it your gene+Method.png
*note maximum likelihood trees will take a long time to build, Neighbor joining is faster
*BOOTSTRAPPING: In terms of your phylogenetic tree, the bootstrapping values indicates how many times out of 100 (in your case) the same branch was observed when repeating the phylogenetic reconstruction on a re-sampled set of your data.
How to build trees using MEGA (by Max Haase '17): http://haasegen564s17.weebly.com/mega.html (note old version of MEGA used) *if you are looking for example files to use in MEGA you can find these files HERE. Try the CRAB rRNA file as an Example.
Alternative ways to build a tree:
Align using Clustal Omega: https://www.ebi.ac.uk/Tools/msa/clustalo/
Then paste the alignment into:
Simple Phylogeny: https://www.ebi.ac.uk/Tools/phylogeny/simple_phylogeny/
Homolog Finding using Ensemble
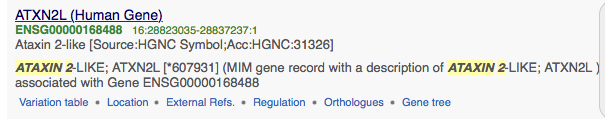
sample Ensemble file...see where the Orthologues link is?
Ensemble directions:
1. Search with your exact gene/protein name in humans.
2. Click ORTHOLOGS in left menu
3. Now select the model organisms at the minimum. To do this you need click Configure This Page (left menu)
This will allow you to select specific species you want to download. Click on only what you want in your tree.
4. Click Download Orthologs (button is just below word Orthologs under your protein name) THEN > Select FASTA > Select Unaligned Sequences > Download
5. Check your desktop for the .fasta file
1. Search with your exact gene/protein name in humans.
2. Click ORTHOLOGS in left menu
3. Now select the model organisms at the minimum. To do this you need click Configure This Page (left menu)
This will allow you to select specific species you want to download. Click on only what you want in your tree.
4. Click Download Orthologs (button is just below word Orthologs under your protein name) THEN > Select FASTA > Select Unaligned Sequences > Download
5. Check your desktop for the .fasta file
This is a phylogenetic tree for the CCR5 protein. This tree was drawn using the BLOSUM62 and the Neighbor Joining method.
Phylogeny
Generate a Tree: In order to generate a phylogenetic tree from the alignment, (1)the similarity between sequences must be determined. (2) Once similarity scores have been calculated by either of these methods, a tree is drawn. Several different methods can be used to draw the tree. Try Maximum Likelihood and Neighbor Joining methods that are found in MEGA.
1. Formatting sequences: First you obtain protein sequences from different organisms of interest (i.e. your homologs)(minimum of all of the model organisms 5+ works best). Next you put these sequences into annotated FASTA format (example: >Human CCR5). See example on Max Haase's Website for how he formatted this Word file with his homologs. How you format this page is SUPER important. Weird spacing and such will cause havoc. To circumvent this issue, save your Word file as a .txt file (this removes all of the weird spacing you might have).
2. Align sequences & build simple tree in ClustalWOmega: You will align all of the sequences up with each other using ClustalWOmega. Now Click the button to Send to SIMPLE Phylogeny in ClustalWOmega. This will build a simple tree. You will see that you now have to annotate your .fas or .txt file with the names of your species for the tree to look nice. So you have to go back to this .fas file and add the species name after the >. For example, Change: >ENSTRUP00000043038 to >Homo sapiens
Do your alignment all over again with this annotated file to produce a nice final and basic tree.
1. Formatting sequences: First you obtain protein sequences from different organisms of interest (i.e. your homologs)(minimum of all of the model organisms 5+ works best). Next you put these sequences into annotated FASTA format (example: >Human CCR5). See example on Max Haase's Website for how he formatted this Word file with his homologs. How you format this page is SUPER important. Weird spacing and such will cause havoc. To circumvent this issue, save your Word file as a .txt file (this removes all of the weird spacing you might have).
2. Align sequences & build simple tree in ClustalWOmega: You will align all of the sequences up with each other using ClustalWOmega. Now Click the button to Send to SIMPLE Phylogeny in ClustalWOmega. This will build a simple tree. You will see that you now have to annotate your .fas or .txt file with the names of your species for the tree to look nice. So you have to go back to this .fas file and add the species name after the >. For example, Change: >ENSTRUP00000043038 to >Homo sapiens
Do your alignment all over again with this annotated file to produce a nice final and basic tree.

example of an alignment in MEGA for Ataxin-2
3. NOW try the program MEGA.
You will have to download this to your desktop to use.MEGA DOWNLOAD
4. How to build trees using MEGA (by Max Haase '17): http://haasegen564s17.weebly.com/mega.html (note old version of MEGA used) *if you are looking for example files to use in MEGA you can find these files HERE. Try the CRAB rRNA file as an Example.
5. Put your trees on your website: Save your trees to your website. Go to IMAGE > SAVE File as .PNG. Then you upload the tree by inserting an Image widget. Discover the ways you can visualize the trees (Rectangular, Straight and Curved or Circle) by using the left Tree button at the just below Subtree on your tool bar.
Explain on your website what the differences are between Neighbor Joining, Maximum Likelihood and Average distance are.
You will have to download this to your desktop to use.MEGA DOWNLOAD
4. How to build trees using MEGA (by Max Haase '17): http://haasegen564s17.weebly.com/mega.html (note old version of MEGA used) *if you are looking for example files to use in MEGA you can find these files HERE. Try the CRAB rRNA file as an Example.
5. Put your trees on your website: Save your trees to your website. Go to IMAGE > SAVE File as .PNG. Then you upload the tree by inserting an Image widget. Discover the ways you can visualize the trees (Rectangular, Straight and Curved or Circle) by using the left Tree button at the just below Subtree on your tool bar.
Explain on your website what the differences are between Neighbor Joining, Maximum Likelihood and Average distance are.
Websites used:
Homology:
Entrez Gene: https://www.ncbi.nlm.nih.gov/gene
Ensemble: https://useast.ensembl.org/index.html
Phylogeny Programs:
MEGA: http://www.megasoftware.net
ClustalWOmega: https://www.ebi.ac.uk/jdispatcher/msa/clustalo
Entrez Gene: https://www.ncbi.nlm.nih.gov/gene
Ensemble: https://useast.ensembl.org/index.html
Phylogeny Programs:
MEGA: http://www.megasoftware.net
ClustalWOmega: https://www.ebi.ac.uk/jdispatcher/msa/clustalo
Pasting HTML code into Weebly
How to paste stuff from a Word file into your web pages.
Type as you normally would in Word. Now whatever you want to post on your site you need to copy and paste into a new word file. After you have done that. Save as a Webpage (this is under the File tab). Now your file will have a .htm at the end.
After you publish it you might see that the font size it too big. If this happen you have got back to Word and format in a different Font size and re-save it again like you did above.
Type as you normally would in Word. Now whatever you want to post on your site you need to copy and paste into a new word file. After you have done that. Save as a Webpage (this is under the File tab). Now your file will have a .htm at the end.
- Then go up to View>HTML Source
- The file will now look like code.
- Go to Weebly and insert a Custom HTML block on to your page.
- Copy and paste this code into the box after clicking Edit Custom HTML.
- Publish.
After you publish it you might see that the font size it too big. If this happen you have got back to Word and format in a different Font size and re-save it again like you did above.
Maximum Likelihood
In this method, an initial tree is first built using a fast but suboptimal method such as Neighbor-Joining, and its branch lengths are adjusted to maximize the likelihood of the data set for that tree topology under the desired model of evolution.
Neighbor Joining
The Neighbor Joining method essentially takes the similarity scores generated by percent identity or BLOSSUM and uses these scores to determine which species are most closely related. It then calculates the branch lengths (or amount of change that occurred after the two species diverged) and draws a tree. An example of the Neighbor Joining tree tree for CCR5 is shown here. Unlike the Average Distance Tree shown on the phylogeny page, branches are all different lengths [3].
Average Distance
The Average Distance method uses the similarity scores to determine which species are most closely related and joins them with equal branch lengths. This assumes that both species have diverged equally from the common ancestor [3]. An example of a tree created using Average Distance is shown on Anna-Lisa's phylogeny page.
In this method, an initial tree is first built using a fast but suboptimal method such as Neighbor-Joining, and its branch lengths are adjusted to maximize the likelihood of the data set for that tree topology under the desired model of evolution.
Neighbor Joining
The Neighbor Joining method essentially takes the similarity scores generated by percent identity or BLOSSUM and uses these scores to determine which species are most closely related. It then calculates the branch lengths (or amount of change that occurred after the two species diverged) and draws a tree. An example of the Neighbor Joining tree tree for CCR5 is shown here. Unlike the Average Distance Tree shown on the phylogeny page, branches are all different lengths [3].
Average Distance
The Average Distance method uses the similarity scores to determine which species are most closely related and joins them with equal branch lengths. This assumes that both species have diverged equally from the common ancestor [3]. An example of a tree created using Average Distance is shown on Anna-Lisa's phylogeny page.